Synthetic DNA Nanotechnology – reprogramming a familiar molecule
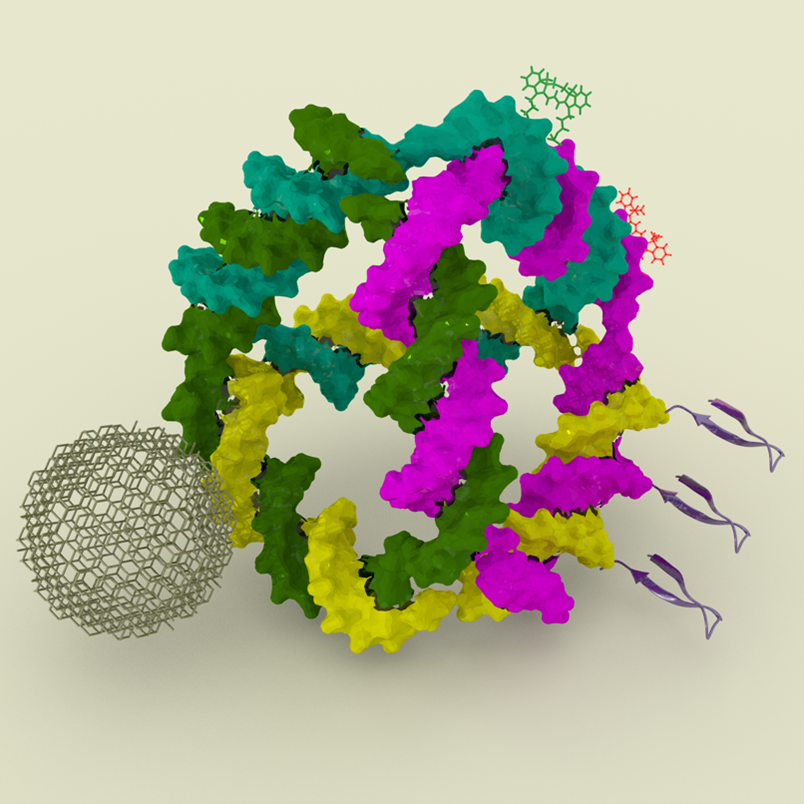
Abstract: Synthetic DNA nanotechnology facilitates the design and fabrication of nanoscale particles and devices with diverse applications. Leveraging a growing toolkit of DNA self-assembly methods, it is possible to construct both two- and three-dimensional structures ranging from nanometer to micron scales. The biophysical and biochemical properties of DNA — combined with its compatibility with various organic and inorganic nanoparticles and its predictable base-pairing rules — have made it an ideal material for single-molecule studies, photonics, plasmonics, synthetic biology and healthcare applications. In this work, we present our efforts in developing DNA-based platforms to organize inorganic and organic nanoparticles and biosensors precisely. We investigate how these DNA scaffolds can control the positioning and orientation of nanoparticles to enhance their photophysical properties. Additionally, we explore the behavior of DNA nanostructures when introduced into mammalian cell cytosol, a critical step toward creating biocompatible delivery systems for therapeutic and diagnostic purposes. Finally, we will discuss our recent efforts in building gene-encoded DNA nanoparticles, a promising advancement in the development of targeted delivery systems.
Website: https://www.mathurnanolab.com/