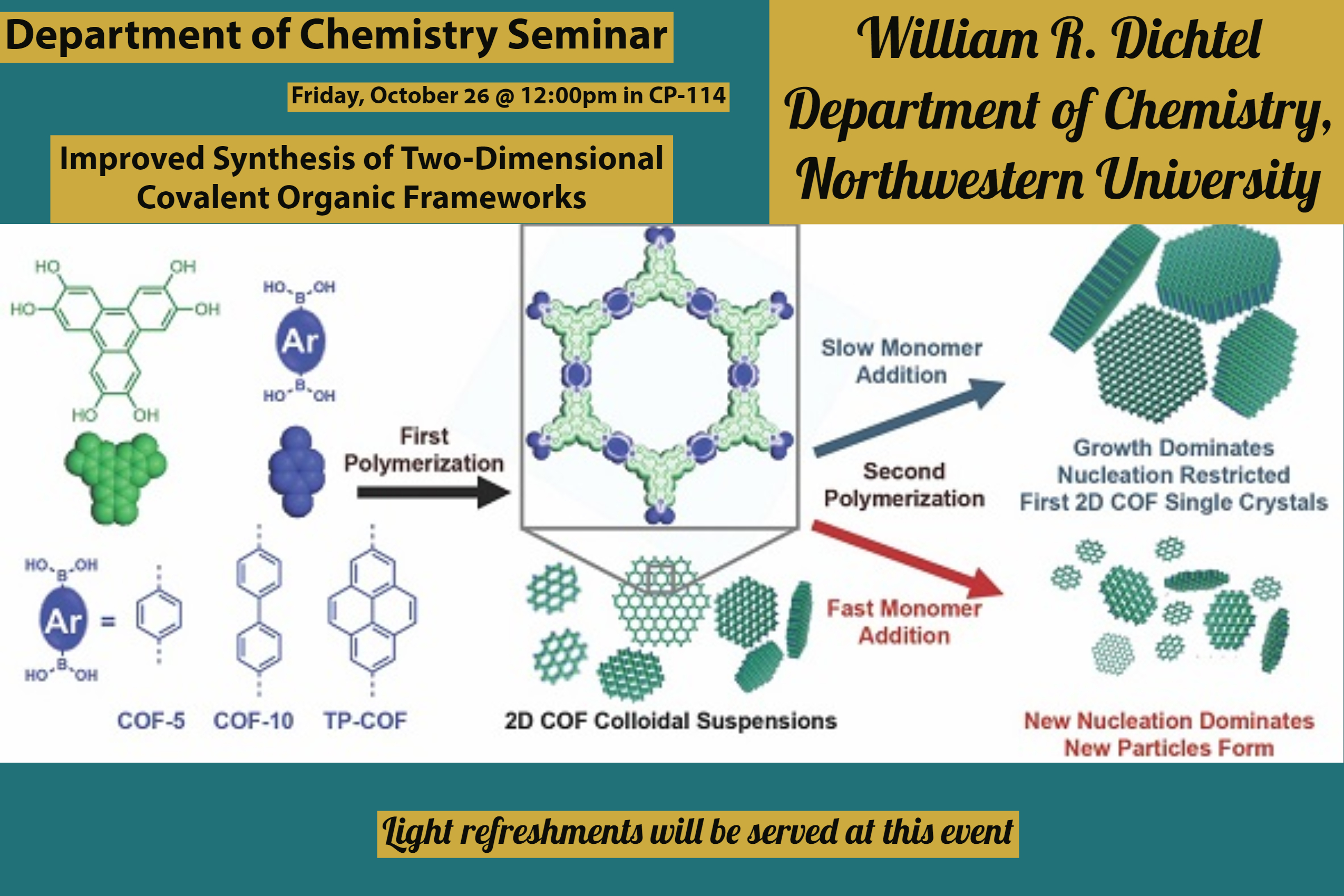
Abstract: Although the conversion of oleaginous biomass to the fatty acid methyl esters (FAMEs) that constitute biodiesel is a mature technology, feedstock availability issues as well as challenges stemming from the high oxygen content of FAMEs have limited the widespread application of biodiesel. Consequently, attention has shifted to processes capable of catalytically deoxygenating oleaginous biomass to afford fuel-like hydrocarbons. Deoxygenation via decarboxylation/decarbonylation (deCOx) represents a promising alternative to the hydrodeoxygenation (HDO) processes typically employed to achieve this transformation, as deCOx does not necessitate the high pressures of hydrogen and the problematic sulfided catalysts required by HDO.
To date, the majority of deCOx reports involve Pd or Pt catalysts, the cost of which may be prohibitive. However, Ni-based catalysts have been shown to be capable of affording comparable results to precious metal-based formulations [1]. Recently, we have observed that the promotion of Ni with other earth-abundant metals – such as Cu – leads to considerable improvements in activity, selectivity and resistance to coking [2]. Results of Temperature Programmed Reduction (TPR) and X-ray Photoelectron Spectroscopy (XPS) measurements suggest that these improvements can be attributed to the ability of the aforementioned metal promoters to improve the reducibility of Ni. This results in an increased amount of Ni0, which is believed to be the active phase in the deCOx reaction.
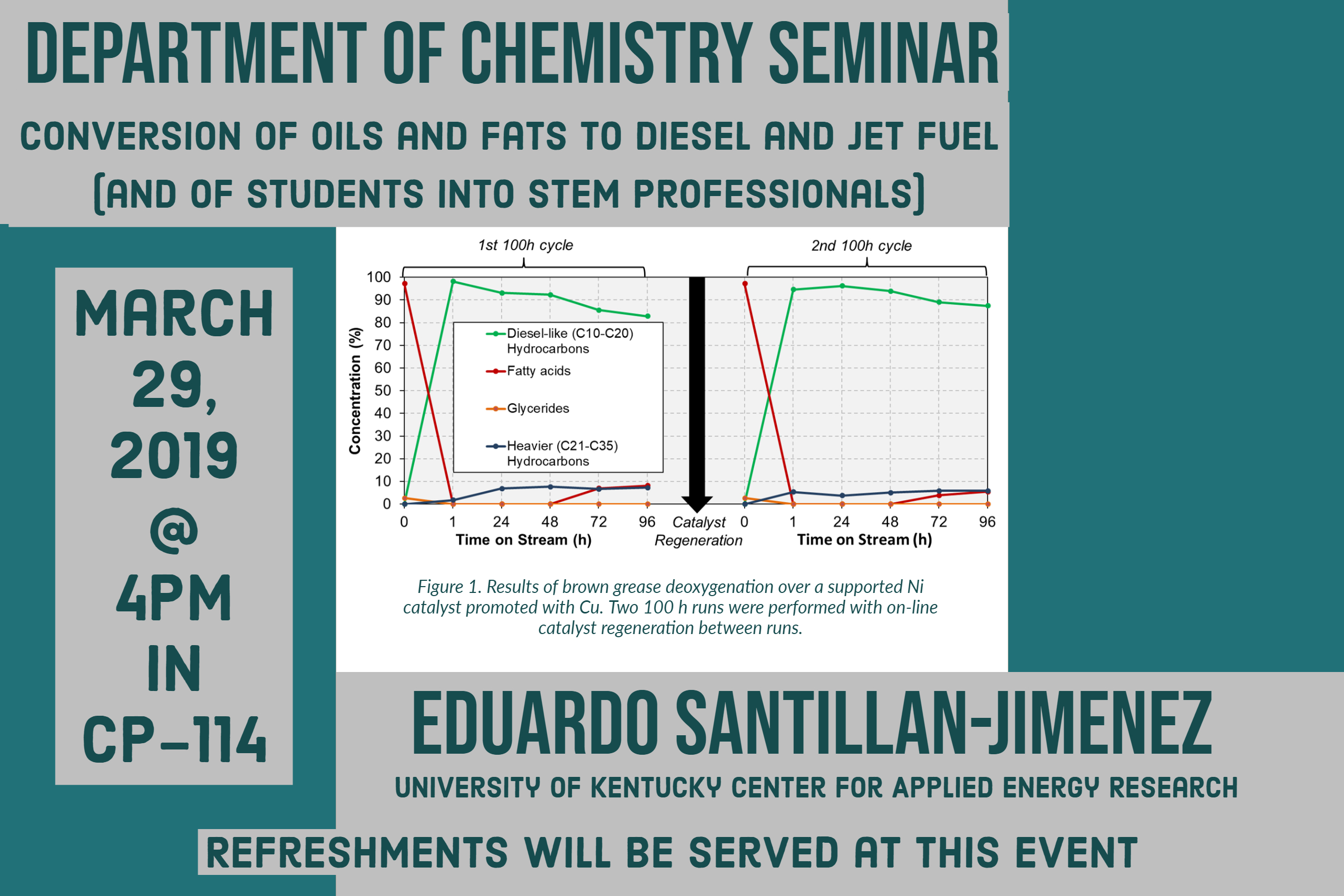
Ni catalysts promoted in this manner afford remarkable results in the conversion of a wide variety of model, waste and/or highly unsaturated lipids – including tristearin, triolein, yellow grease, brown grease, hemp seed oil and algal FAMEs – to fuel-like hydrocarbons [3-6]. Indeed, using a fixed-bed reactor operated using industrially-relevant reaction conditions, close to quantitative yields of diesel-like hydrocarbons are obtained. In addition, as shown in Figure 1, a catalyst employed has displayed remarkable stability and recyclability in a run comprising two 100 h time on stream cycles [5].

Mentoring has been identified as an effective tool not only for attracting and retaining students from groups traditionally underrepresented in STEM disciplines, but also for improving their academic performance. However, additional benefits could be obtained by housing mentoring initiatives in research centers as opposed to in traditional academic departments. Therefore, a mentoring initiative based at the University of Kentucky Center for Applied Energy Research is striving to test this hypothesis [7]. Recently, providing the participating students access to international research opportunities has become a focus of this mentoring program.
References
[1] T. Morgan, D. Grubb, E. Santillan-Jimenez, M. Crocker. Top. Catal., 2010, 53, 820.
[2] R. Loe, E. Santillan-Jimenez, A.F. Lee, M. Crocker, et al. Appl. Catal. B: Environ., 2016, 191, 147.
[3] E. Santillan-Jimenez, R. Pace, T. Morgan, C. McKelphin, M. Crocker, et al. Fuel, 2016, 180, 668.
[4] E. Santillan-Jimenez, R. Loe, M. Garrett, T. Morgan, M. Crocker. Catal. Today, 2018, 302, 261.
[5] R. Loe, M. Maier, M. Abdallah, R. Pace, E. Santillan-Jimenez, M. Crocker, et al. Catalysts, 2019, 9, 123.
[6] R. Loe, K. Huff, M. Walli, R. Pace, Y. Song, E. Santillan-Jimenez, M. Crocker, et al. Catalysts (IN PRESS).
[7] E. Santillan-Jimenez, W. Henderson. 124th American Society for Engineering Education Annual Conference Proceedings, 2017, Conference Paper ID #17681.




 Michael Harmata was born in Chicago in 1959. He attended St. Michael the Archangel grammar school, Thomas Kelly High School, and the University of Illinois-Chicago, where he received his AB degree in chemistry with a math minor in 1980, working in the labs of Jacques Kagan and graduating with honors and highest distinction and all that great stuff that doesn’t matter anymore. He earned his PhD under the tutelage of Scott E. Denmark at the University of Illinois in Urbana, Illinois in early 1985, working on carbanion-accelerated Claisen rearrangements. He then did an NIH postdoc with Paul A. Wender at Stanford University where he performed some of the first work on the synthesis of the neocarzinostatin chromophore. He began his independent career at the University of Missouri-Columbia in 1986, where he is now the Norman Rabjohn Distinguished Professor of Chemistry. He has been contributed significantly to the areas of (4+3)-cycloaddition reactions, benzothiazine chemistry, pericyclic reactions of cyclopentadienones, chiral molecular tweezers, silver-catalyzed chemistry, and more.
Michael Harmata was born in Chicago in 1959. He attended St. Michael the Archangel grammar school, Thomas Kelly High School, and the University of Illinois-Chicago, where he received his AB degree in chemistry with a math minor in 1980, working in the labs of Jacques Kagan and graduating with honors and highest distinction and all that great stuff that doesn’t matter anymore. He earned his PhD under the tutelage of Scott E. Denmark at the University of Illinois in Urbana, Illinois in early 1985, working on carbanion-accelerated Claisen rearrangements. He then did an NIH postdoc with Paul A. Wender at Stanford University where he performed some of the first work on the synthesis of the neocarzinostatin chromophore. He began his independent career at the University of Missouri-Columbia in 1986, where he is now the Norman Rabjohn Distinguished Professor of Chemistry. He has been contributed significantly to the areas of (4+3)-cycloaddition reactions, benzothiazine chemistry, pericyclic reactions of cyclopentadienones, chiral molecular tweezers, silver-catalyzed chemistry, and more. Abstract:
Abstract: